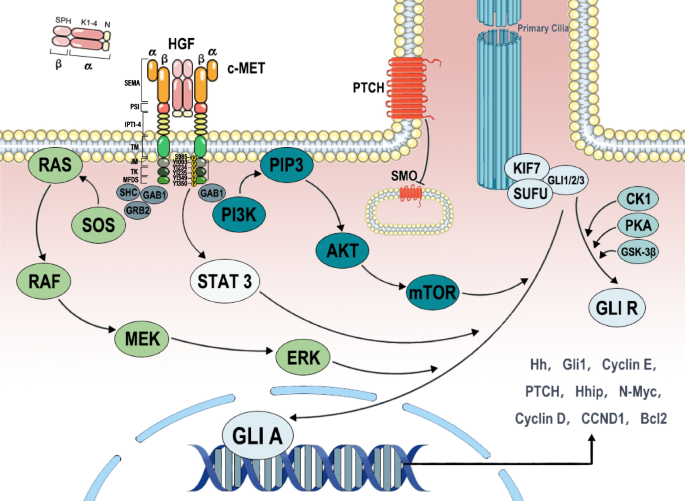
MET的突变在癌症的发生中起着至关重要的作用,而Hedgehog (Hh)通路在细胞分化和肿瘤干细胞的维持中也起着重要作用。传统的化疗药物主要针对肿瘤内的大多数细胞群,而不是肿瘤干细胞。因此,在短暂的缓解期后,肿瘤常常复发。此外,肿瘤干细胞的特异性靶向忽视了其他肿瘤细胞恢复干细胞和获得耐药的可能性。因此,目前仅针对HGF/c-MET轴和Hh通路的药物在特定类型的癌症中仅显示出中等疗效。越来越多的证据表明,这两种途径不仅在癌症中发挥重要作用,而且通过其自身配体的分泌,对单靶点治疗的耐药发展产生重大影响。在这篇综合综述中,我们分析和比较了Hh通路对HGF/c- met驱动肿瘤模型中肿瘤微环境(TME)的潜在影响,以及不同细胞类型之间的相互作用。此外,我们进一步证实了双途径联合治疗作为MET依赖性癌症治疗的关键靶点的潜力和必要性。
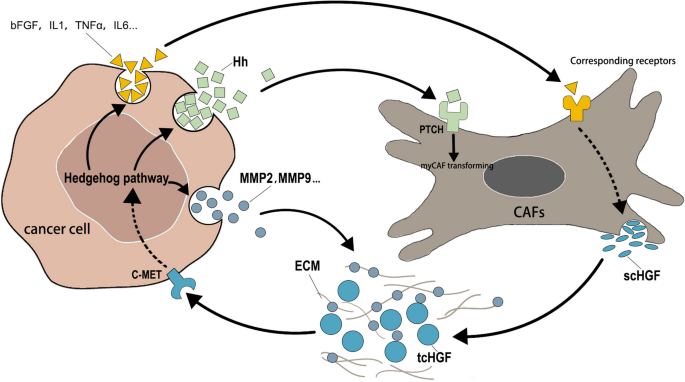
图书馆的src="http://zrsww.com/news/show/226203/ / / e.video-cdn.net/v2/embed.js " > 视频摘要 HGF/c-MET是在生长发育过程中起重要作用的重要分子轴。HGF,即肝细胞生长因子,代表配体,而c-MET指的是它的受体,一种受体酪氨酸激酶。该信号通路在促进细胞增殖、迁移、分化、血管生成甚至干细胞自我更新等过程中发挥着重要作用,具有精确的时空表达特征。这些功能在胚胎发育、多器官形成和损伤修复过程中至关重要[1,2,3]。然而,c-MET受体的过度激活或突变可能导致不受控制的细胞增殖、逃避凋亡和上皮-间质转化。这些异常现象可驱动癌细胞的恶性转化和播散,从而促进肿瘤的形成和进展[4,5,6,7]。因此,HGF/c-MET信号通路已成为研究癌症发展机制和确定潜在治疗靶点的关键领域。 Hedgehog (Hh)通路是另一种在生长、发育和癌症中起关键作用的信号通路[8]。Hh通路参与胚胎发育、器官形成和组织修复过程,对维持正常细胞增殖、分化和模式形成至关重要[9]。该通路的突变可导致多种遗传性疾病,包括前脑畸形、Greig头多指畸形、Pallister-Hall综合征、Carpenter综合征等,并与多种癌症的发生和进展密切相关[10]。研究表明,在许多肿瘤类型中,Hh通路的异常激活可以促进癌细胞的增殖、存活和侵袭能力,并有助于肿瘤干细胞的形成和维持。此外,Hh通路的异常激活与肿瘤的关键特征相关,如血管生成、免疫逃避和耐药性。因此,研究Hh通路的调控机制并制定相关的治疗策略已成为癌症研究的重要课题,为癌症治疗提供了新的方向和可能性。 癌相关成纤维细胞(Cancer-associated fibroblasts, CAFs)是肿瘤微环境(tumor microenvironment, TME)的关键组成部分,通过促进血管生成、侵袭和转移来促进肿瘤进展[11]。HGF/c-MET轴和Hh通路在肿瘤细胞与CAFs相互作用中起关键作用。已经证明Hh通路促进CAF激活并招募CAF来调节细胞分化[12]。激活的CAFs通过分泌HGF刺激肿瘤细胞中的HGF/c-MET轴,进一步增强肿瘤的干性、侵袭和转移[13]。然而,靶向其中的单一途径仅在有限数量的肿瘤中显示出中等疗效。长期使用不可避免地会产生耐药性[14,15,16,17]。因此,本文旨在探讨这两种信号通路在与肿瘤细胞和CAFs相互作用的背景下相互作用的机制。 本文综述了目前对肿瘤细胞和CAFs中HGF/c-MET轴和Hh通路以及它们之间相互作用的了解。此外,我们将探索同时靶向HGF/c-MET轴和Hh通路作为癌症治疗的合理方法的潜力。我们相信,同时靶向这两种途径有望更有效地抑制肿瘤生长,并对癌症患者的耐药和无进展生存产生积极影响。 随着癌症治疗探索的深入,HGF/c-MET轴已成为一个重要的靶点。HGF/c-MET通路在多种肿瘤中被异常激活,在肿瘤发生、增殖、转移、血管生成、干细胞等多种生物学过程中发挥关键作用。c-MET是HGF的受体,由MET原癌基因编码。它是由furin蛋白酶水解其前体形成的,形成由胞外α-链和跨膜β-链由二硫键连接组成的二聚体结构[18]。HGF以单链前体(scHGF)的形式分泌,并经过细胞外蛋白水解加工形成活性双链形式(tcHGF),其由α-重链(69 kDa)和β-轻链(34 kDa)组成。scHGF和tcHGF都能与c-MET结合;然而,只有经过处理的tcHGF才能激活c-MET信号转导。最近的研究表明,在tcHGF中,高亲和力α-链和低亲和力β-链都含有c-MET的结合位点。α-链单独结合不激活c-MET受体;相反,它作为c-MET的结合域,促进β链的低亲和力结合,最终导致c-MET活化[19]。在癌症背景下,异常的HGF分泌和激活,加上与过表达、扩增和选择性剪接密切相关的MET基因突变,导致HGF/c-MET轴的异常激活。大量证据表明,这种失调是多种癌症的肿瘤发生和进展的驱动因素,包括肾乳头状细胞癌(PRCC)[20]、头颈部鳞状细胞癌(HNSCC)[21]和结直肠癌(CRC)[22]。这些作用是通过一系列下游信号通路介导的,包括但不限于PI3K/AKT、Ras/MAPK和Wnt/β-catenin。 在生长发育过程中,c-MET主要在上皮源性细胞中表达,而HGF主要作为形态原在间质源性细胞中表达,并通过旁分泌和/或自分泌机制在器官形成、细胞极性决定、细胞迁移、组织损伤修复、特化上皮组织维持等过程中发挥重要作用[23,24,25]。在肿瘤细胞中,已经确定了特定的met相关突变以提高催化效率。然而,体外研究表明,这些突变不足以独立诱导细胞转化。此外,即使在正常水平表达野生型MET的细胞中,HGF的过表达也被证明能有效促进肿瘤的发生。这一现象在转基因小鼠模型中得到了验证,强调了活性HGF对c-MET激活的至关重要[26]。虽然一些肿瘤细胞能够产生HGF及其剪接变体,从而通过自分泌和旁分泌信号调节肿瘤进展,但基质来源的HGF仍然是c-MET持续激活不可或缺的元素[27,28,29,30]。此外,更多的研究表明,一些肿瘤基质细胞,如caf和肿瘤相关巨噬细胞,是旁分泌机制中HGF表达和释放的主要来源[31]。虽然大多数间质细胞在早期对肿瘤细胞有一定的限制能力,但最终会促进恶性肿瘤的生长、侵袭和转移。它们可以通过旁分泌使HGF高浓度存在于肿瘤间质中,从而促进肿瘤细胞的恶性行为[32,33]。 TME中各种基质细胞释放的高水平HGF是基质和原发肿瘤之间恶性串扰的关键参与者。HGF在关键的TME组分CAFs中大量表达,而其受体c-MET在肿瘤细胞中高表达[34]。活化的CAFs可以加速机体内多种恶性肿瘤的生长[35]。大量实验研究表明,一些恶性肿瘤细胞在体内表现出侵袭性,但大多数肿瘤细胞在体外不侵袭基质凝胶。只有当与CAFs共培养或将其条件培养基掺入胶原蛋白凝胶中时,癌细胞才会表现出对胶原蛋白凝胶的侵袭潜力。在这种情况下,由cas分泌的最重要的细胞因子之一是HGF[36,37,38]。有证据表明,HGF是通过肿瘤-基质相互作用赋予肿瘤细胞侵袭性生长潜能的关键分子之一[39]。最近的体内实验进一步证实了这一观点。在小鼠肿瘤模型中,使用lrat - cre转基因小鼠删除来自肝星状细胞(HSC)衍生的cas或来自肿瘤细胞源的MET的HGF,观察到肿瘤侵袭性和大小显著降低[40]。这些观察结果也在乳腺癌[41]、结直肠癌[42]、胆管癌[43]等多种癌症类型中得到证实。 在细胞外基质(ECM)中,存在着一个复杂的蛋白质和大分子网络。这种ECM作为一种生物屏障,保护肿瘤免受免疫系统反应和外部治疗剂的影响。它还可以作为各种生长因子的储存库。HGF已被证明能与多种ECM蛋白紧密结合,包括血栓反应蛋白-1 (TSP-1)、纤维连接蛋白、层粘连蛋白、I型胶原蛋白、硫酸肝素、蛋白聚糖和基底膜组分[39]。通过这些与ECM的关联,HGF被隔离在其中,在不同的动态位置形成信号分子的浓度梯度。这些梯度诱导肿瘤细胞侵袭和迁移。肿瘤细胞和基质细胞都可以分泌修饰ECM的蛋白酶,包括基质金属蛋白酶(MMPs)、丝氨酸蛋白酶、组织蛋白酶、崩解素和金属蛋白酶(ADAMs),以及ADAMTS(一种带有血栓反应蛋白基元的崩解素和金属蛋白酶)家族的成员[44,45]。这些蛋白酶,如HGFA、基质酶和MMP-2,不仅将scHGF转化为其成熟的形态tcHGF,而且还促进结合在ECM上的HGF的释放,导致自身刺激[46]。ECM释放的某些生长因子,如转化生长因子α (TGF-α),可以通过激活其表皮生长因子受体与c-MET相互作用,在没有配体的情况下触发信号转导[47]。这与细胞因子和生长因子形成了一个复杂而动态的网络。 此外,邻近cas分泌的HGF显著增强肿瘤细胞的干性,调节肿瘤细胞的代谢。常见的干细胞相关分子如CD44可以将HGF招募到细胞膜上,促进其传递到c-MET并调节其活性[48]。癌症干细胞(CSCs)可以产生HGF的诱导剂,包括IL-1β、PDGF、TNF-α、bFGF和前列腺素E2 (PGE2)。这些诱导剂促进间充质干细胞的转化,维持cas的表型,导致cas释放HGF增加。ECM中高浓度的HGF作用于肿瘤细胞的c-Met受体,进一步增强肿瘤的侵袭性生长。 摘要。 介绍 癌症中的HGF/c-MET轴 癌症中的Hh通路 HGF和Hh环在癌细胞和CAF之间的作用 针对HGF/c-MET轴和Hh通路的单一抑制的临床状况 Hh通路和HGF/c-MET轴在met突变癌中的临床影响 结论 未来的发展方向 数据和材料的可用性 参考文献。 作者信息 道德声明 # # # # # Hh通路不仅在上皮-间质转化过程中与HGF/c-Met协同作用,而且作为形态因子在调节TME中发挥重要作用[49]。Hh通路在进化中高度保守,对胚胎发育、组织修复和纤毛功能有重要影响[50,51]。Hh最早是在果蝇中发现的,由于Hh位点的突变导致突变体角质层上形成方向相反的小突带,类似于Hh的棘,因此被命名为“Hh”。在哺乳动物中,Hh有三个亚型:Sonic Hh (Shh)、Indian Hh (Ihh)和Desert Hh (Dhh),它们在进化中高度保守,在身体的不同部位发挥重要作用。其中,SHH在肿瘤中的研究最为广泛[52]。与需要细胞外加工才能成熟的HGF不同,Hh蛋白在细胞内加工。翻译后,Hh前体首先进行自催化裂解,形成含有内部肽硫酯的前体。然后,在哺乳动物中,胆固醇与c端的硫酯发生反应,而n端则被Hh酰基转移酶(HHAT)棕榈酰化(黑腹果蝇中的瘦Hh)。在经历了这两个重要的脂质修饰后,Hh被紧紧地固定在细胞膜上。随后,分派同源物1 (DISP1)作用于与Hh共价连接的脂质,并将Hh与可溶性载体信号肽- cub结构域和egf样结构域2 (SCUBE2)包装并释放Hh[53,54]。分泌的Hh可在组织间产生浓度梯度,在脊椎动物的肢芽中,其距离可达300 μm[55]。此外,Hh可被Hh相互作用蛋白(HHIP)结合,抑制其功能[56]。补丁同源物1 (PTCH1)是Hh通路中的另一个重要分子,也是抗性-结节分裂(RND)家族的成员,负责特异性接受脂质修饰的Hh[57]。 在经典的Hh信号通路中,Hh通过与具有12个跨膜结构域的跨膜蛋白Patched (Ptc)结合,与其受体一起传递信号。在果蝇中,这种蛋白被称为Ptc,而在哺乳动物中,PTCH有两种类型,主要是PTCH1蛋白和PTCH2蛋白。PTCH1被认为在这一途径中起主要作用。在Hh缺失的情况下,PTCH1通过抑制Smoothened (SMO)抑制Hh通路的激活。这种作用可能是通过ptch1介导的胆固醇从细胞质膜外排实现的,这是SMO激活所必需的[58]。当脂质修饰的Hh与PTCH1结合时,PTCH1不再抑制SMO, SMO可被PKA、CK1α和GRK2磷酸化和激活。在Kif3A和β-阻滞蛋白的帮助下,SMO转运到纤毛膜,在那里它促进Gli的核定位,并通过抑制PKA、GSK-3β和ck1介导的Gli磷酸化依赖的泛素化和降解激活该途径。在Hh通路下游,Cyclin D、Cyclin E、Myc、Gli1、PTCH、BCL2、VEGF、Fox、IL-1β、IL-3、IL-6、TNFα等多个基因可通过转录调控。 如前所述,包装的Hh是可以释放的,研究表明,在初级纤毛尖端释放的Hh与在基侧膜释放的Hh的扩散距离有显著差异[50]。释放的Hh可以作用于表达Ptch的细胞,从而激活Hh通路。这在TME中肿瘤细胞与其他细胞的相互作用中起着重要作用。有研究表明,即使在Hh通路未发生突变的情况下,上皮细胞(包括结肠癌、胰腺癌、卵巢癌、肺癌、口腔癌等癌症)产生的Hh配体也会激活周围基质细胞中的Hh通路,从而创造有利于肿瘤生长的微环境,间接促进肿瘤生长[59,60,61]。此外,在前列腺癌和胰腺癌等癌症中,旁分泌比自分泌发挥更大的作用[62,63]。例如,Hh通路抑制剂环巴胺在体外对前列腺癌细胞株22Rv1的生长没有明显的抑制作用,但在异种移植试验中显示出抑制作用[64,65]。这一观察结果可能表明,Hh通路抑制剂的作用并不局限于肿瘤细胞本身,而是应该关注整个TME。在肿瘤细胞内,更多Hh蛋白的产生是通过独立于SMO的非规范激活途径发生的。反过来,TME中的细胞以源自肿瘤细胞的Hh依赖方式维持激活,这导致Hh信号的抑制对肿瘤细胞的影响有限,但对基质细胞很敏感[66]。 CAFs是TME中最重要的细胞之一。在肿瘤形成的早期阶段,各种成纤维细胞类型和间充质祖细胞可以被肿瘤细胞募集和/或激活,影响肿瘤炎症、纤维化和癌症进展等多种生物学行为。这些相似但独特的成纤维细胞类型统称为CAFs[67]。基于其表型特征,CAFs目前主要分为两大类:肌成纤维CAFs (myCAFs),表达高水平的α-平滑肌肌动蛋白(α-SMA)和成纤维细胞激活蛋白(FAP);炎性CAFs (iCAFs),具有分泌特征并调节炎症[68]。虽然通过劫持肿瘤细胞的生长和发育途径驱动致癌突变是已知的,并且对癌症研究至关重要,但几乎每种类型的基质细胞都具有在特定情况下支持癌细胞的能力。因此,CAFs提供的旁分泌和有丝分裂信号可能影响几乎任何肿瘤发生阶段的不同类型的肿瘤,并参与从异常增殖到侵袭、迁移和耐药的过程。 如前所述,caf通过分泌HGF在激活癌细胞中的Met信号传导中起着至关重要的作用。同样,已经证实,HGF在CAFs中的表达通常高于正常成纤维细胞(NFs),并且在许多肿瘤中都有记录。这些来自CAFs的HGF分子通过旁分泌信号以2:2的方式激活邻近肿瘤细胞中的c-Met。有趣的是,最近的研究,利用低温电子显微镜的结构洞察,揭示了单个HGF分子可以桥接两个相对的c-MET分子,导致它们的激活,第二个HGF分子进一步稳定这种结合并增强c-MET的激活[19]。HGF激活的两个c-MET分子在Tyr1234和Tyr1235上发生同二聚化和自磷酸化,随后诱导残基Y1349和Y1356的自磷酸化。这种激活涉及Src同源-2 (SH2)结构域、磷酸酪氨酸结合(PTB)结构域和met结合结构域(MDB),招募各种下游分子,包括GRB1、GRB2、SHC、PI3K和STAT3,从而通过多种下游信号通路增强Hh通路活性[69](图1)。 图1 HGF/c-Met轴通过下游信号激活Hh通路,包括MAPK、PI3K/AKT和STAT3等。HGF的kringle结构域(K1-K4)结构激活c-Met,而丝氨酸蛋白酶同源结构域(SPH)和N端结构域(N)增强受体结合。c-Met的细胞外部分由SEMA结构域、丛蛋白信号整合素(PSI)结构域和丛蛋白和转录因子(IPT1-4)结构域的四个免疫球蛋白样区域组成。胞内部分包括近膜(JM)结构域、酪氨酸激酶(TK)结构域和c端多功能对接位点(MFDS)。JM结构域S985和Y1003的磷酸化导致泛素化介导的c-Met降解,而TK结构域Y1234和Y1235的自磷酸化通过触发MFDS中Y1349和Y1356的磷酸化来上调途径活性。MFDS的磷酸化主要用于募集下游蛋白 丝裂原活化蛋白激酶(MAPK)通路是c-Met激活的重要下游信号通路[70]。c-MET招募Grb2(结合位点Y1356)后,可激活Raf/MEK/ERK信号级联。最终,GLI1不仅可以被ERK2激活(在残基S102、S116和S130处)[71],还可以激活下游激酶,包括MSK1/2和pp90RSK,以调节GLI蛋白活性[72]。而c-Met中Y1356的磷酸化也可以激活下游的PI3K/AKT/mTOR信号通路。在食管癌中,mTOR可以通过S6K1激活GLI1磷酸化(在Ser84处),导致GLI1与SUFU分离,并增加其转录活性[73]。这一概念在前列腺癌和软骨肉瘤的研究中得到支持[74,75]。然而,在神经母细胞瘤研究中,未观察到明显的调节作用,提示肿瘤异质性可能决定了信号通路激活的差异[76]。此外,mTOR可以通过抑制4EBP1激活Hh通路,促进细胞增殖,这在小鼠小脑和成神经管细胞瘤中得到证实[77]。此外,活化的AKT不仅使GSK-3β失活,还调节Hh和Wnt通路。AKT可磷酸化β-catenin的Ser552位点,增加其转录活性,诱导GLI1和GLI2的表达[78]。STAT3与磷酸化的c-Met结合,进行自我磷酸化,并易位到细胞核,增加与肿瘤发生相关基因的转录[79]。STAT3不仅经常与GLI1和GLI2形成转录复合物,结合到它们的锌指结构域促进转录,还可以直接增强慢性淋巴细胞白血病细胞中GLI1的表达[80]。此外,最近的研究表明,STAT3对于成神经管细胞瘤中smo依赖的信号传导也是必不可少的[81]。 当Hh通路过度激活时,下游基因,包括Hh、IL-1β、IL-6、TNF-α、bFGF和FOXF1,直接或间接被转录激活。这通过旁分泌信号导致caf和HGF的表达增加。值得注意的是,FOXF1还可以作为HGF的转录激活因子,进一步促进其转录[82]。通过比较口腔鳞状细胞癌患者衍生的CAFs与正常口腔成纤维细胞中Hh通路的激活情况发现,肿瘤细胞分泌的SHH是CAFs中Hh通路的关键激活因子,而在正常口腔成纤维细胞中,尽管SHH和GLI1表达,但该通路仍然相对不活跃[59]。一些研究表明,旁分泌Hh信号可以驱动CAFs向myCAFs表型分化[33,40]。myCAFs产生大量的ECM,导致高度交联的ECM形成物理屏障,压缩血管组织并阻碍药物传递。这种现象在胰腺癌、胆管癌、肝癌、头颈部鳞状细胞癌等癌症中尤为突出。此外,密集的ECM导致营养物质消耗和缺氧环境,阻碍了免疫细胞的激活。缺氧环境还可诱导MET原癌基因的过表达和c-MET的活化,使HGF信号通路扩增,激活缺氧诱导基因的转录,最终增加肿瘤的干性。此外,Hh通路激活的肿瘤细胞可以依靠下游的MMPs诱导剂,包括MMP2和MMP9,降解胶原IV、胶原VII和糖蛋白,从而修饰ECM并释放结合到基质上的HGF。这促进了肿瘤的侵袭,并利用ECM作为营养来源。当Hh通路被抑制时,它会导致myCAFs减少和iCAFs增加。有报道称,iCAFs是分泌HGF的主要亚群,进一步促进肿瘤细胞内Hh的激活,以补偿Hh抑制剂的抑制作用,最终增加肿瘤的耐药性[83]。 肿瘤中的Warburg效应导致肿瘤细胞的代谢变化,并通过产生乳酸改变TME。HGF和活化的肌成纤维细胞样CAFs (myCAFs)也有助于这种代谢转变。HGF可以增强几种癌症中葡萄糖转运体GLUT1和GLUT4的表达,增加糖酵解和营养消耗[21,27,84]。此外,myCAFs合成的大量ECM有助于缺氧和乳酸生成。在这种TME中,乳酸的积累既可以通过NF-κ b依赖的方式增加caf中HGF的表达,也可以通过肿瘤细胞中ERK/p90RSK通路诱导MMP表达来修饰ECM[85,86]。如前所述,乳酸还可以激活IL-6/STAT3和Wnt/β-catenin等信号通路[87],最终导致Hh通路的激活,形成恶性循环。此外,在一项研究中,作者观察到在胰腺导管腺癌小鼠模型中,当他们抑制HGF/C-MET和Hh通路时,另一途径的代偿激活。这进一步证实了关于这两种通路之间存在恶性串扰的结论[88](图2)。 图2 癌细胞和CAF之间的HGF和Hh环 肿瘤募集CAFs涉及HGF的分泌,HGF反过来通过肿瘤细胞内的C-MET激活下游信号转导。这个级联最终导致Hh信号通路的激活。在Hh通路的下游,一方面,Hh、IL-1β、IL-6、TNF-α、bFGF和FOXF1会被诱导,促进CAFs内HGF的生成。另一方面,Hh的细胞间传递也可以激活CAFs内的Hh通路,从而促进其转化为肌成纤维细胞样CAFs (MyCAFs)。这种转化导致细胞外基质(ECM)的产生增加,诱导干性,促进侵袭性,并产生抵抗免疫细胞细胞毒性和药物毒性的屏障。此外,随着Hh通路的激活,下游事件包括基质金属蛋白酶(MMPs)的产生,其修饰ECM。 如前所述,无论MET基因组是否发生变化,HGF/c-MET轴的激活在各种恶性肿瘤中都有报道。这种激活与肿瘤的干性和耐药性显著相关[89]。目前,c-MET抑制剂主要应用于非小细胞肺癌(NSCLC)、小细胞肺癌(SCLC)、乳头状肾细胞癌(PRCC)、胃癌(GC)、乳腺癌(BC)、肝细胞癌、黑色素瘤、多形胶质母细胞瘤(GBM)等具有高频率met相关突变(尤其是外显子14跳变(METex14)和扩增)的肿瘤[90,91,92,93]。针对HGF/C-MET的靶向治疗目前包括选择性C-MET酪氨酸激酶抑制剂(TKIs)、非选择性C-MET TKIs、抗C-MET单克隆抗体和抗HGF抗体。虽然许多针对HGF/C-MET轴的临床试验正在进行或已经完成,但大多数结果并未显示完全的肿瘤消退和靶标接合的明确证据。此外,在目前的临床试验中,已经观察到接受C-MET抑制剂的患者获益中等,并且随着治疗的延长,癌症的进展和靶标耐药是不可避免的[69]。 目前,广泛应用的Hh通路抑制剂主要可分为两类。一类包括靶向SMO (Smoothened)的抑制剂,SMO是Hh通路的关键组成部分。这类药物包括环巴胺(通过与SMO胞外和跨膜结构域内的口袋结合来抑制Hh信号转导)、TPB15[94]、vismodegib、saridegib和Sonidegib。与临床转译成功有限的环巴胺相比,后三种药物表现出更强的稳定性,已进入临床实践或获得监管部门批准上市[95]。此外,如前所述,甾醇是SMO激活所必需的,一些阻断细胞内胆固醇合成的药物,如他汀类药物和伊曲康唑,可用于阻止SMO激活[96]。由于长期靶向后SMO位点突变导致耐药的发生,以及非经典途径介导的GLI激活,第二类靶向Hh通路的药物是该通路下游的GLI转录因子抑制剂,如三氧化二砷(ATO)[97]、glabrescione B (gla)[98]、hpi[99]、JK184[100]、GANT-58、GANT-61[64]。然而,与靶向HGF/c-MET类似,靶向Hh通路的临床益处仅限于少数癌症,如BCC和MB。此外,vismodegib联合吉西他滨的临床试验表明,尽管Hh信号活性显著降低,但vismodegib的加入并没有提高总体缓解率或延长患者生存时间[101]。这些一致的失败表明Hh信号反应的复杂性及其在癌症中的不同作用,Hh信号通路作为一个高度保守的信号网络,其代偿机制和复杂性以及在不同癌症中的作用有待进一步探索。 因此,单独靶向HGF/C-MET轴或Hh通路作为对抗癌症干细胞和耐药的策略似乎是不够的。考虑到前面描述的复杂的细胞相互作用和这两种途径的代偿性质,针对这两种途径的联合治疗可能会产生更好的结果。 毫无疑问,考虑到HGF/c-MET和Hh通路在肿瘤细胞和CAFs中的相互作用,以及细胞内的串扰和自分泌机制,同时靶向HGF/c-MET和Hh通路可能会产生更理想的效果。 由于非小细胞肺癌(NSCLC)在TKI治疗中不可避免地出现耐药性,Hh通路和c-MET信号的高频激活最初引起了人们对c-MET和SMO双重抑制的关注。利用“计算机药物再利用”和结构分析方法,glesatinib和foretinib被鉴定为能够同时阻断c-MET和SMO的化合物。这在tki耐药NSCLC的细胞实验和异种移植肿瘤模型中得到了验证[102]。随后的研究也证明了格沙替尼在METex14和I型MET抑制剂耐药肿瘤模型和患者中的持续有效性。值得注意的是,在克唑替尼治疗后复发的metex14阳性和MET Y1230H突变患者中,该药仍然有效[103]。在I期临床试验中,格沙替尼单药治疗NSCLC和晚期实体瘤met激活突变人群的客观缓解率(ORR)为30.0%[104]。在另一项晚期实体瘤的临床试验中,格沙替尼联合厄洛替尼或多西他赛的ORR分别为1.8%和12.0%[105]。在随后的二期试验中,晚期或转移性NSCLC患者接受格沙替尼单药治疗,肿瘤组织中MET激活突变组的无进展生存期(PFS)为3.95个月,ORR为10.7%,而肿瘤组织中MET基因扩增组的PFS为4.84个月,ORR为15% (NCT02544633)。同样,在一项针对晚期肝癌患者的I/II期多中心研究中,福替尼的PFS为4.2个月,ORR为22.9%[106]。在一项针对乳头状肾细胞癌的II期临床试验中,福替尼的PFS为9.3个月,ORR为13.5%[107]。在一项针对三阴性复发或转移性乳腺癌患者的II期研究中,总体部分缓解率加上疾病稳定率为38%,中位缓解持续时间分别为4.4个月和5.4个月[108]。这些结果表明,c-MET和SMO的双重抑制对未选择的晚期实体瘤患者有中等疗效,对MET突变的晚期患者有希望。 在目前的肿瘤学研究中,针对CSCs的靶向治疗的重点主要集中在几种信号通路上,包括Wnt、Hh、Notch、Hippo以及与它们密切相关的NF-κB、MAPK、PI3K和EGFR[109]。治疗策略主要旨在抑制特定癌症类型中最相关的途径,或针对异质肿瘤细胞群中适当的分子实体[110]。尽管长期以来人们认识到这些途径在肿瘤发生中的重要性,并且从单细胞研究中获得了越来越多的见解,这些研究揭示了TME中CAF的亚型,并且更详细地描述了它们参与由HGF/c-MET和Hh等途径介导的干细胞和耐药,但仍然缺乏针对不同CAF群体的靶向治疗方法。此外,截至目前,还没有公开的临床试验结果确定同时靶向Hh通路和HGF/c-MET轴。大多数涉及Hh通路抑制剂的临床试验主要集中在评估其疗效和将这些抑制剂整合到现有的化疗方案中,包括嘧啶核苷类似物。然而,这些研究往往忽略了肿瘤细胞内以及肿瘤细胞与TME中其他细胞类型之间发生的代偿信号,导致在许多实体肿瘤中引入Hh通路抑制剂后的结果不理想。相比之下,许多基于细胞的小鼠实验已经证明了肿瘤细胞和CAFs之间的相互作用在肿瘤异质性中的关键作用,一些使用SMO抑制剂和非特异性TKIs的临床试验取得了良好的结果。此外,如前所述,针对SMO和MET的特异性TKIs的出现进一步支持了这一现象,因为它们都对晚期实体瘤(包括那些对常规治疗有耐药性的实体瘤)表现出广泛的抑制作用。我们的研究旨在提供见解,为什么单一靶向Hh途径,尽管是一个关键的干性途径,产生次优的治疗效果和不可避免的耐药,当TKIs单独使用时。我们通过总结肿瘤细胞内主要c-MET和Hh通路之间复杂的串扰,Hh通路激活对TME和CAFs的下游影响,以及SHH和HGF在基质-肿瘤相互作用背景下作为桥梁的作用来实现这一目标。本研究对肿瘤细胞内原代c-MET和Hh通路之间的串扰进行了全面分析。此外,我们还研究了Hh通路激活对TME和CAFs的下游影响。此外,我们还探索了Hh和HGF作为介质在肿瘤细胞和基质之间相互作用中的作用。根据细胞间通讯和信息传递的观点,我们的研究解决了为什么单独靶向Hh通路导致治疗效果不佳以及为什么单独使用TKIs不可避免地导致耐药的问题。重要的是,我们的研究提供了更深层次的机制理解,特别是表型研究,并为癌症治疗的多靶点方法提供了新的方向。 虽然福替尼和格沙替尼的出现及其令人鼓舞的临床试验结果暂时解决了在c-MET驱动的肿瘤中同时抑制c-MET和SMO的空白,但仍然缺乏针对c-MET和SMO的更特异性和更有效的药物的临床试验数据。临床试验应迅速启动,特别是在肿瘤表现MET突变和间质增殖。此外,值得注意的是,目前的临床研究主要集中在晚期肿瘤。然而,由于在疾病进展过程中,即使在同一肿瘤类型和不同细胞群中也可能出现肿瘤内异质性[111],双途径抑制可能对早期met驱动的肿瘤产生更有利的结果。此外,还需要对不同MET突变肿瘤中Hh通路激活的细胞内机制进行更深入的研究。研究细胞内信号通路之间的直接相互作用对于指导未来的治疗策略至关重要。 最后,目前Hh通路抑制的重点主要集中在靶向SMO上。然而,越来越多的证据指向SMO靶向导致的突变。GLI是Hh通路中的关键转录因子,许多旨在抑制GLI的药物面临着与毒性相关的挑战,阻碍了它们的临床转化。值得注意的是,像伊曲康唑和ATO这样已经上市的药物,由于它们的分布,可能会在特定的实体肿瘤中引起不可避免的毒性问题。因此,有必要进行更多的研究,以抑制GLI作为Hh通路中的转录因子。这一研究途径有可能发现更安全、更有效的治疗策略。此外,考虑到MET基因的频繁突变以及MET对HGF的依赖,深入研究开发针对cas的特异性HGF抑制剂是非常有必要的。这条研究路线有可能加深我们对TME的理解,并为癌症治疗领域的创新、靶向治疗铺平道路。 ccDownload: /内容/ pdf / 10.1186 / s12964 - 023 - 01333 - 8. - pdf