
汉坦病毒(HTN)是汉坦病毒科的成员。它是一种分节型负链病毒(sNSVs)。它引起肾综合征出血热,包括发烧、血管出血和肾功能衰竭。这种疾病是世界上最严重的出血性疾病之一,由于其高死亡率,它是一个主要的公共卫生问题。汉滩病毒RNA依赖的RNA聚合酶复合体(RdRp)参与病毒RNA转录和复制,以维持该病毒的生存和传播。因此,它是抗病毒药物开发的主要靶点。干扰HTN病毒RdRp内切酶结构域的内溶性“夺帽”反应是一种特别有吸引力的药物发现方法。HTN病毒的RdRp内切酶结构域具有金属依赖性的催化活性。我们针对这种金属依赖的酶活性,利用计算机方法,即分子对接,分子动力学模拟,预测吸收,分布,代谢,排泄,毒性(ADMET)和药物相似性研究,确定可以结合和破坏这种内切酶活性的抑制剂。对接研究表明,peramivir和ingavirin化合物可以有效地与锰离子结合,并与该蛋白的其他活性位点残基结合。分子模拟还显示这些配体与HTN RdRp活性位点的稳定结合。模拟分析表明,它们与HTN病毒核酸内切酶结构域的活性位点锰离子和氨基酸残基持续接触。这项研究将有助于更好地了解HTN和相关病毒。
汉坦病毒是正汉坦病毒属汉坦病毒科病毒的RNA病毒,具有负链片段基因组,属于布尼亚病毒目[1]。蝙蝠、鼩鼱、鼹鼠和啮齿动物是汉坦病毒的哺乳动物宿主[2,3]。汉坦病毒有几种类型,有些是非致病性的,有些是致病性的,可引起人类严重疾病。这些疾病包括肾综合征出血热(HFRS)和汉坦病毒心肺综合征(HCPS)[4]。这些致病性病毒可通过不同途径向宿主传播。其中包括通过雾化啮齿动物排泄物传播[5],2020年阿根廷也报告了人与人之间的传播[6],也可能通过输血传播[7]。
不同类型的汉坦病毒分布在世界各地;它们也可以被分为旧大陆和新大陆汉坦病毒。汉滩病毒、首尔病毒、多布拉瓦-贝尔格莱德病毒等旧世界病毒会引起HFRS疾病。HCPS疾病是由新世界汉坦病毒(如sinnombre病毒(SNV)、New York-1病毒和Andes病毒)引起的[8]。蛋白尿、血尿、急性肾损伤都是HFRS的症状,死亡率高达15%[9]。在亚洲和欧洲,Dobrava、Hantaan、Seoul和Puumala病毒是引起HFRS的最常见血清型。每年的病例数在6万至15万之间,与该地区的啮齿动物种群有关[10,11]。HCPS是一种由汉坦病毒引起的呼吸衰竭和弥漫性间质水肿为特征的发热性疾病[12]。
它是肾综合征出血热的主要直接病因,以发热、血管出血和肾功能衰竭为特征。这些情况统称为肾综合征出血热(HFRS)[13]。该疾病与埃博拉出血热一样是世界上最严重的出血性疾病之一,由于其高死亡率,是一个主要的公共卫生问题[14,15]。每年的病例数在6万到15万之间[10,11]。HTN具有三段负链RNA基因组,并被包膜。病毒RNA聚合酶、两种重要的膜表面糖蛋白(G1和G2)和核衣壳蛋白(N)分别由基因片段L、M和S编码[16,17]。汉坦病毒主要在动物和人的内皮细胞、肾小球和上皮细胞内感染和复制[18]。HTN病毒进入宿主细胞时,首先与细胞表面受体整合素β- 1,2和3结合,然后通过网格蛋白介导的内吞作用内化到宿主细胞中,也有报道HTN在内化宿主细胞时也利用宿主细胞的动力蛋白[19]。
RNA依赖性RNA聚合酶(RNA-dependent RNA polymerase, RdRp)是汉滩病毒基因组转录和复制的主要聚合酶,是汉滩病毒基因组转录和复制的主要蛋白,分子量为250 ~ 280 kDa。HTN的RdRp除了具有转录酶和复制酶活性外,还具有其他几种酶活性。RNA解旋酶功能是RdRp的额外活性之一,另一方面,它缺乏盖帽和校对所需的酶活性[20]。正链和负链RNA病毒都有一个在RdRp中保守的结构聚合酶结构域,HTN RdRp蛋白也是如此。HTN RdRp核心域的整体结构包括一个右手折叠,可分为三个子域:拇指、手指和手掌域。HTN RdRp有4个基序A-至D-,其聚合酶功能位于棕榈子结构域的c基序中[21]。
一种独特的夺帽核酸内切酶结构域与流感病毒的核酸内切酶结构域有一定的相似性[22]。cap-snatching活性对于启动RdRp的RNA聚合酶功能至关重要。为了开始其vRNA的转录,HTN病毒需要mRNA寡核苷酸,它使用mRNA寡核苷酸作为引物来启动病毒转录过程。这种mRNA引物是由cap-snatching domain提供的,cap-snatching domain从宿主细胞mRNA和核酸内切酶domain一起从宿主细胞mRNA中抓取mRNA帽,并在复制位点切割和加工宿主mRNA供病毒使用。然后将得到的10-15个碱基的带rna帽的引物用于病毒mRNA转录初始化[23]。HTN病毒聚合酶从宿主mRNA上夺取mRNA帽,然后通过其RdRp内切酶结构域将其切割,并将其转移到RdRp的聚合酶结构域。其他病毒如登革病毒利用其盖帽酶在宿主体内启动病毒基因组合成[24]。
汉坦病毒这种RNA导向的RNA聚合酶- l蛋白cap- snatch核酸内切酶结构域通过将宿主mRNA转移到主RdRp结构域开始病毒RNA转录,实现了cap- snatchandcutting宿主mRNA的重要功能。从图1可以看出,汉滩病毒核酸内切酶结构域活性位点含有两个具有催化活性的二价金属锰离子。其中一个Mn2+离子由His36、Asp97、Glu110和Val111配位,第二个Mn2+离子由活性位点残基的Glu54和Asp97配位[23]。
图1
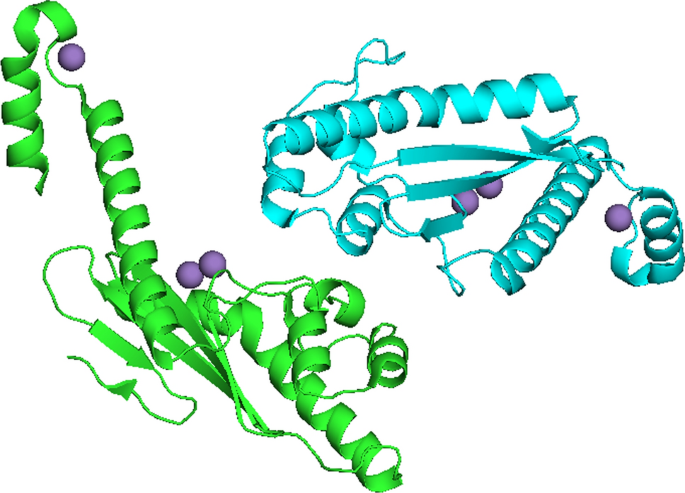
汉滩病毒RNA依赖的RNA聚合酶- l蛋白cap-snatching核酸内切酶结构域蛋白结构,活性位点上有两个锰离子,末端位置上有一个锰离子,这是稳定性和核酸酶活性所必需的(PDB ID 5IZE) [23]
因此,这种内切酶结构域是发现抗HTN病毒感染新药的一个有希望的靶点。在这项工作中,通过各种计算方法探索了汉滩病毒的内切酶结构域。利用分子对接和分子动力学模拟等方法对多种化合物和抗病毒药物进行筛选。类似地,其他的计算机方法也被用来鉴定潜在的抗病毒分子,这些分子可以有效地螯合锰金属离子,并与活性位点残基相互作用。这些抗病毒分子破坏了RdRp催化活性位点残基内Mn2+金属离子的结合,完全抑制了RdRp的功能。
从PDB ID号为5IZE的RCSB蛋白数据库中获得了汉坦RdRp l蛋白的cap- snatchedase结构域的晶体结构[23]。该结构的分辨率为1.70 ?。RdRp的这种抢帽内切酶为二聚体形式,二聚体在RCSB网站上被命名为“Biological Assembly-1”和“Biological Assembly-2”,因为该蛋白的两个单体相同,结构和功能相似,所以我们选择了“Biological Assembly-1”单体并下载了其蛋白PDB结构进行计算研究。
我们进行了深入的文献研究,以选择和检索能够靶向HTN RdRp酶内切酶结构域的抑制化合物。HTN RdRp在其活性位点含有两个Mn+2离子,这对该内切酶结构域的催化活性至关重要。选择这里使用的化合物的标准是它们螯合和参与活性部位离子的潜力。因此,这些化合物可以有效地参与和阻断HTN RdRp酶的催化活性。以前,已经报道了几种与不同病毒靶酶具有金属螯合活性的化合物,例如(流感病毒及其内切酶结构域抑制剂Xofluza https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6336199/)。基于这些观察,我们搜索了几个学术在线数据库。PubChem数据库SMILES搜索选项用于进一步探索化合物。选择对数据库中几种病毒具有抗病毒活性的化合物及其衍生物进行对接研究。基于这些抗病毒化合物结构的一些相关合成化合物也从研究文章中检索到。此外,我们还利用了其他几种由不同基团合成并在文献中描述的金属螯合化合物,它们具有靶向这类病毒蛋白靶点的潜力。根据这些标准,我们仔细地检索和选择化合物并将其纳入我们的研究。选用ChemDraw Ultra 12.0软件绘制并制备化合物结构。本研究中使用的所有化合物的化学结构可以在(补充图1)中看到。
通过Molecular Operating Environment (MOE-2015.10)软件对我们的靶蛋白HTN RdRp l -蛋白cap- snatchendonucase domain进行分子对接研究[25]。首先,利用MOE-2015.10内置的“Structure Preparation”模块对蛋白进行分子对接。使用该模块,蛋白质中缺失的原子被添加,蛋白质上的电荷被纠正。蛋白质氨基酸之间的链间断裂键也得到了纠正。电荷和氢也被添加到蛋白质上,然后它被3d质子化。通过MOE中的“site - finder”模块选择HTN RdRp l蛋白cap- snatchendonucase domain的活性位点。我们的蛋白的活性位点及其两个催化金属离子的氨基酸残基列于(表1)中。我们研究期间通过MOE 2015.10对接的参数设为:
1.
位置设置为“三角形匹配器”
2.
细化设置为“刚性受体”
3.
评分功能(两个)设置为“London dG Scoring”
4.
而在分子对接期间的姿势设置为每个化合物20个姿势。
表1氨基酸序列与meHTN RdRp Endo活性部位的tal离子核酸酶域
在分子与靶蛋白对接之前,配体结构的能量也被最小化[15,26,27]。
计算机ADME和毒性研究包括小分子(药物)从胃肠道的吸附,其在生命系统内的分布和代谢,其排泄,以及药物对生物体的毒性预测,可通过几种计算工具确定[28]。我们利用ADMET- sar在线服务器,它可以确定这些小分子的ADMET性质。通过对接研究确定的这些表现最好的小分子的药物样性质预测,通过Swiss-ADME在线服务器进行。该服务器预测的一些类药参数是分子量和拓扑表面积(TPSA)参数,如果预测的这些值对于分子来说最小且更小,那么这些分子就有可能被开发为药物。其他与药物相似的性质是氢键受体和供体的数量以及分子中存在的杂原子(如硫、氮等),如果这些性质存在于分子中,则认为它们更像药物。
薛定谔戴斯蒙用于进行相互作用分析和评价的分子动力学模拟。使用Maestro的“System Builder”来配置所有系统[29]。所有三个确定的顶击配合物都是能量最小化的,并放置在一个正交方框中,缓冲距离为10 ?。使用Builder工具构建了溶剂化水浸MD-Simulation系统。本实验采用TIP3P模型进行溶剂化。通过加入适量的反离子来中和模拟系统。通过在模拟面板中加入0.10 M的钠离子和氯离子来维持等渗状态。在模拟之前,遵循预先定义的平衡程序。MD模拟是在约1.013 bar的环境压力和300 K的温度下进行的,持续时间为100 ns[30,31]。
摘要。
介绍
材料与方法
结果与讨论
结论
数据可用性
参考文献。
作者信息
道德声明
# # # # #
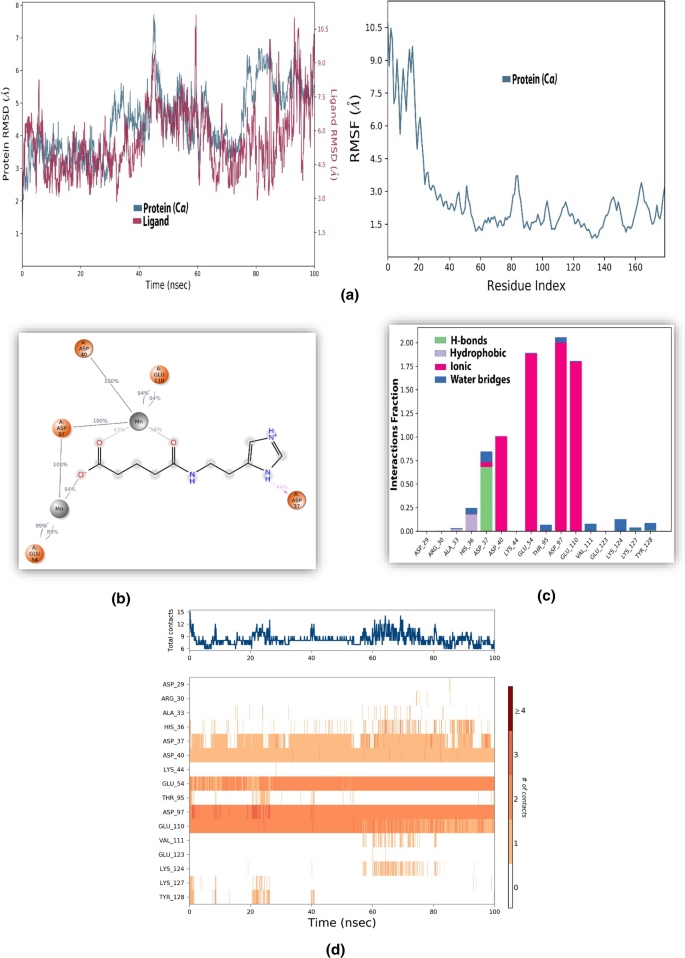
在对接研究中表现良好、RMSD较低、结合自由能(s-scores)适宜、与锰离子和HTN RdRp核酸内切酶结构域活性位点氨基酸残基相互作用次数最多的分子将在本节中进行讨论。
2 ' -脱氧-2 ' -氟胞苷是抗癌药物吉西他滨的类似物。2 ' -脱氧-2 ' -氟胞苷也有报道对SARs-CoV-2具有抗病毒活性[32]。在我们的计算对接研究中,这个小分子显示出与HTN病毒RdRp内切酶结构域的几种相互作用。它与锰离子和参与酶活性的活性位点氨基酸残基以及协调这些金属离子的氨基酸结合并相互作用。它的对接s值或结合亲和能为- 9.21 kcal/mol。该化合物与蛋白质活性位点有7种相互作用。该化合物的嘧啶环与活性位点氨基酸发生两次相互作用。一种是嘧啶环羰基氧与Val113的氢键作用,另一种是芳烃-阳离子与活性位点的Lys124氨基酸的相互作用。该化合物的氟化核糖环参与5种相互作用,该核糖环上的羟基与Lys127、Lys124形成氢键,其中一个-OH也与活性位点的锰离子发生螯合接触。它的第二个锰离子也与核糖环环氧原子发生金属接触。此外,Lys124还与这个环氧原子发生了氢键。该配体-酶复合物的RMSD为0.897 ?。
它也是一种抗病毒药物,是一种3-羟基单羧酸,属于环戊醇-环戊酮、乙酰胺和胍类化合物家族,对流感病毒有活性[33]。与酶活性位点发生7次相互作用,并螯合Mn+2离子。该化合物环戊醇环上的羟基与锰离子螯合,并与帕拉米韦的乙酰胺羰基氧原子发生金属接触作用,而第二个锰离子也被与第一个锰离子螯合的同一个羟基螯合。该羟基与活性位点的Glu110氨基酸形成氢键。乙酰胺部分所连接的碳原子也与His36发生了芳烃- h相互作用。环戊醇上的羧基与Lys124和Lys127形成两个h键相互作用。对接s-score为- 8.14 kcal/mol, RMSD为1.50 ?。
英格韦林也是一种抗病毒药物,用于各种病毒性呼吸道感染[34]。它的结构包含一个羧基、一个咪唑环和一个乙胺基,位于其链结构的中间。英格韦林还显示出与HTN RdRp核酸内切酶活性位点的最佳相互作用。它与锰离子和它的氨基酸残基发生了6次相互作用。英格韦林的乙胺基羰基氧与锰离子螯合并相互作用。乙酰胺所连接的碳原子与His36发生了芳烃-氢相互作用,而英格韦林的羧基与Glu110、Lys124和Lys127活性位点氨基酸发生了三次相互作用。对接s-score为- 8.74 kcal/mol, RMSD为1.60 ?。
该化合物是一种羧酸,其结构中有一个苯基环和一个酮基。与锰离子有良好的螯合作用,与HTN病毒RdRp核酸内切酶结构域活性位点氨基酸有良好的氢键作用。在对接分析中发现了5个键的相互作用。酮(羰基)基氧原子螯合活性位点的锰离子并与之接触。该化合物的羧基、羟基和羰基与Glu110、Lys124和Lys127蛋白活性位点形成3个h键相互作用。其对接s-score(结合亲和能)为?8.13 kcal/mol,对接过程中配体-蛋白复合物的RMSD为1.91 ?。
皮莫地韦也是一种抗病毒药物,对甲型流感病毒(IAV)的RdRp有活性[35]。本对接研究也有效靶向了HTN的RdRp内切酶结构域,并与该酶发生了6次相互作用。活性位点的锰离子的有效螯合作用被注意到,并与活性位点的氨基酸结合。该化合物结构复杂,含有多个环状结构和官能团。在这个化合物中,只有羧基螯合了单个锰离子和氮原子。该氮还与活性位点的Glu54氨基酸结合并与之形成氢键。该化合物的嘧啶环与His36氨基酸发生了两个芳烃-芳烃型相互作用,而与嘧啶环相连的氟原子与Lys124氨基酸形成了一个氢键。该配体的对接s值为- 11.1 kcal/mol, RMSD值为1.25 ?。
雷替格拉韦是另一种用于治疗HIV感染的抗病毒药物。它的靶点是HIV整合酶,一种含有金属离子的金属蛋白,如HTN RdRp内切酶金属离子。HIV整合酶是将HIV病毒基因组整合到宿主基因组中的必需酶,而雷替格拉韦抑制了病毒基因组整合的这一步骤[36]。同样,在我们的研究中也注意到这种化合物的有利相互作用。研究发现,雷替重力韦与锰离子和htnrdrp核酸内切酶结构域的活性位点残基均有相互作用。氧基嘧啶环羰基氧与锰离子相互作用,使嘧啶环与活性位点的His36发生芳烃-芳烃型相互作用。雷替格拉韦的两个羧胺官能团与Asp37和Val113氨基酸形成氢键。雷替重力韦的对接s评分为- 9.09 kcal/mol, RMSD值为1.50 ?。
该化合物是一种流感病毒RdRp内切酶抑制剂,是治疗流感感染的有效药物[37]。在本次对接研究中,该药物与锰离子和活性位点残基发生了显著的相互作用。观察到四种相互作用,该分子中存在的几个羰基与金属离子螯合,并与活性位点的Lys124氨基酸形成氢键。其对接s值为- 9.83 kcal/mol。该复合物的RMSD为1.53 ?。
该化合物也是IAV RdRp内切酶结构域的抑制剂。该研究还显示了与HTN内切酶的合适相互作用。氧吡喃环羟基和羰基分别与Glu123和Val113形成氢键。该化合物的磺酰胺基团与HTN RdRp内切酶活性位点的Lys127发生了4次相互作用,有效地结合了锰离子和2个氢键。其对接s值为- 8.41 kcal/mol, RMSD值为1.14 ?。
金葡霉素是一种从链霉菌中分离出来的抗菌化合物。该小分子的对接s值为- 6.09 kcal/mol, RMSD值为1.05 ?。这个分子中的丙酰胺基团与两个锰离子相互作用。同时,氧化二硫代吡咯环还与其中一个锰离子螯合,并与HTN RdRp核酸内切酶结构域Lys124活性位点氨基酸形成氢键。
该化合物含有一个氧三氮杂环,一个磺酰氧基(-O-SH)和一个二酮(在其三环上含有两个酮基)。该分子与HTN RdRp内切酶也表现出良好的相互作用,并与其活性位点发生6次相互作用。酮羰基氧原子与其中一个金属锰离子相互作用,与Lys124氨基酸形成氢键。第二个锰离子与其中一个环上的氧结合并与之相互作用。而第二酮羰基氧原子与Ala33氨基酸形成氢键相互作用。该化合物的磺酰氧基(-O-SH)也与HTN RdRp内切酶活性位点的Val113氨基酸发生了氢键相互作用。该分子的对接s值为- 7.09 kcal/mol, RMSD值为1.39 ?。
该化合物也是二酮类化合物,先前文献报道对IAV的内切酶结构域有活性[38]。在我们的对接研究中,该化合物有效地与锰离子相互作用,并与该酶的活性位点残基形成氢键。该化合物吡嗪环上的甲氧基氧与活性位点的锰离子相互作用,而吡嗪环上的酮羰基氧也与金属锰离子螯合。该吡嗪环还与Lys124活性位点残基发生了芳烃-阳离子型相互作用。而氟取代苯基则与HTN RdRp内切酶活性位点的Val113氨基酸发生芳烃- h型相互作用。其对接分数为?7.30 kcal/mol,蛋白配体复合物的RMSD值为2.41 ?。
这个化合物含有一个氧三氮杂环,是一个二酮。该分子与HTN RdRp内切酶也表现出良好的相互作用,并与其活性位点发生了5次相互作用。环结构上的甲氧基与一个金属锰离子螯合,活性位点的第二个金属离子与酮羰基氧原子相互作用,该氧原子也与活性位点的Lys124形成氢键。而该化合物的第三个杂环与His36发生了芳烃-芳烃相互作用。在同一环上的酮基也通过其羰基氧原子与HTN RdRp内切酶结构域活性位点的Val113形成氢键。该分子的对接分数为- 9.41 kcal/mol, RMSD值为1.25 ?。
该化合物与前面讨论的化合物相同,但不是两个酮羰基,而是其中一个被羧基取代,其余的结构特征相同。它是一种HIV整合酶抑制剂。在我们的研究中,CHEMBL4435971对其中一个锰离子表现出良好的螯合活性,并通过该化合物环结构上的羰基氧和甲氧基氧原子与锰离子相互作用。该化合物的吡嗪环与His36氨基酸发生芳烃-芳烃型相互作用。同时,该环上取代的酮和羧基也与Val113和Lys124活性位点残基发生了两个氢键相互作用。其对接s-score为- 10.40 kcal/mol, RMSD值为1.33 ?。
这种化合物属于苯酰胺类化合物。该化合物的羧基氧有效地螯合了HTN RdRp核酸内切酶结构域活性位点的Mn+2离子,而苯基环则与Lys127氨基酸进行了芳烃阳离子型相互作用。一个羟基的三羟基苯基环与酶的Val113活性位点残基形成h键。该化合物与活性位点的对接s值为- 11.73 kcal/mol, RMSD值为1.29 ?。
该化合物也是一种甲型流感病毒(IAV)内切酶抑制剂。它是一种羧酸,具有良好的螯合活性。对接结果显示与HTN的RdRp内切酶有5种相互作用。磺酰基氧原子螯合其中一个锰离子。这个磺酰基和另一个氧原子也与Lys124活性位点残基形成氢键相互作用。该化合物结构中的酮官能团氧原子与第二个Mn+2离子螯合,并与取代磺酰基的苯环上的His36发生芳烃-氢键相互作用。该化合物的对接s值为-12.98 kcal/mol, RMSD值为1.78 ?。
上述化合物在二维和三维形式的分子对接过程中得到的对接位姿,以及它们的化学结构和名称列于图2。三维图像由Biovia DS软件绘制,二维对接姿态由内置的MOE-2015-10配体相互作用模块获取。通过ChemDraw Ultra-12.0软件制备化学结构。
在对接研究中,几乎一半的化合物与HTN RdRp核酸内切酶结构域的活性位点表现出合适的相互作用和螯合活性。它们具有良好的对接s值和较低的RMSD值。筛选到的50个配体与HTN病毒RdRp核酸内切酶结构域的分子对接结果见表2。
表2筛选到的主要配体与HTN病毒RdRp末端的分子对接结果核酸酶域
此外,表现最好的三种化合物进行了进一步的计算机计算研究。利用ADMET- sar服务器,还进行了ADMET和药物相似性研究等方法来预测它们在生命系统中的生物利用度、毒性和排泄机制。而这三个主要发现的药物相似性研究是通过SwissADME在线服务器进行的。
对于计算机ADMET预测研究评估,我们选择通过分子对接研究确定的top lead hit,并使用admetSAR[39]服务器。本研究的三种先导化合物均具有良好的人体肠道吸收(HIA),并被列为HIA +。第一个命中的化合物2′-脱氧-2′-氟胞苷的HIA吸收值增强最大,其次是英格韦林,然后是帕拉米韦。这三种化合物都是血脑屏障+,具有良好的血脑屏障吸收值。血脑屏障(BBB)对这三种化合物的吸收也是如此。2′-脱氧-2′-氟胞苷的血脑屏障值增强程度更高,其次是因加韦林,然后是帕拉米韦,均在可接受范围内。当检查这些先导化合物的p -糖蛋白(P-gp)排放估计值时,它们都是非P-gp蛋白抑制剂。同样,这些化合物都没有抑制肾脏有机阳离子转运蛋白,也没有影响其在尿液中的药物分泌活性。
在代谢评价研究中,发现三种先导化合物均不影响细胞色素- p450酶的活性,被列为该类酶的非抑制剂。细胞色素p450酶的非抑制剂表明这些化合物不会干扰细胞色素p450类酶代谢的化合物的生物转化。此外,AMES毒性谱显示这三种化合物是非诱变剂,不会引起或诱发妊娠期先天性胎儿缺陷。在对这些化合物的致癌性研究中,发现它们都是非致癌性的。在口服毒性计算机评价研究中,所有铅化合物都被列为第三类有毒化合物。这一类含有LD50浓度超过500毫克/公斤但低于5000毫克/公斤的化合物,它们是毒性较低的化合物。相比之下,根据美国环境保护署(US-EPA),毒性最大的化合物被列为第一类。2′-脱氧-2′-氟胞苷在大鼠模型中的致死剂量LD50值为2.0673 mol/kg。ingavirin和peramivir的LD50浓度分别为1.9207 mol/kg和2.5219 mol/kg。这些都是高LD50值,这意味着这三种化合物在给药时将被生物系统很好地耐受。所有预测的ADMET性质列在表3中。
表3 ADMET简介in-silico前三名化合物的预测值
前三种先导化合物通过计算机预测研究进一步进行药物相似性测试,以检查这些化合物是否有可能用作对抗HTN病毒的药物。这些研究是通过SwissADME服务器[40]进行的,该服务器通过检查这些化合物的各种化学和结构特征,即拓扑表面积(TPSA)来预测小分子的药物相似特性,拓扑表面积(TPSA)可以定义为分子中表面极性原子的数量。化合物表面极性原子的增加导致TPSA值增加,TPSA值较高的物质是更好的p糖蛋白底物,这意味着它们以更高的速率从细胞中排出,并且具有较低的膜通透性。因此,化合物TPSA值越低,其药性越强。建议TPSA小于等于140 ?2。类药物物质的另一个特点是分子量较低;大多数药物的分子量较小,这增加了它们的吸收。因此,大多数药物都是以尽可能小的分子量制造出来的。但是,稍高的TPSA和分子量也可以被认为是类药,因为FDA已经批准了几种不完全符合Lipinski药物规则的药物[41],但较高的TPSA和分子量会导致药物在gi道的吸收较低,药代动力学特性较差[42]。2′-脱氧-2′-氟胞苷的TPSA值;因加韦林和帕拉米韦分别为110.60 ?2、95.08 ?2和151.03 ?2。分子量分别为245.21 g/mol、225.24 g/mol、328.41 g/mol和281.28 g/mol。所有化合物均具有可接受的TPSA和分子量值,在水中具有良好的溶解度,并且在可耐受范围内具有良好的gi道吸收。这四种化合物都满足并完全遵循利平斯基药物相似规则。我们的顶级先导化合物的所有其他利平斯基药物规则参数见表4。
表4我们前三先导化合物的利平斯基药物规则参数
此外,化合物的药物相似性所依赖的其他物理化学性质,包括化合物中的恢复度、大小、亲脂性、不溶性、极性和柔韧性(化合物中可旋转键的数量),也已在我们的每个顶级化合物的雷达图(图2a-c)中给出。
图2
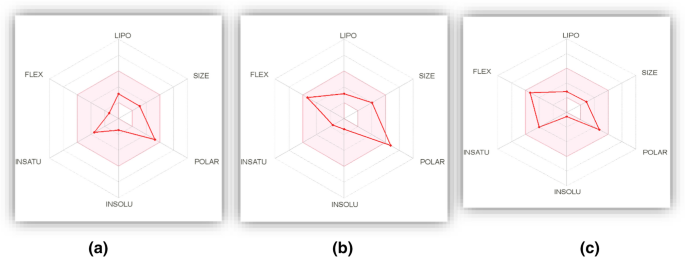
2'-脱氧-2'-氟胞苷的物理化学性质雷达图。b帕拉米韦的物理化学性质雷达图。c ingavirin的理化性质雷达图
此外,进一步的药代动力学分析表明,这些化合物的生物利用度评分为0.55。通常,候选药物需要至少0.10的生物利用度评分才能考虑进一步的药物评价研究。正如swissADME web server预测的那样,这些化合物也具有良好的水溶性[40]。
下图的雷达图(图2a-c)中的彩色空间表示类药物化合物的口服生物利用度的适当物理化学空间。
自由HTN RdRp内切酶的模拟
为了更好地分析HTN RdRp核酸内切酶结构域的相互作用和动力学以及分子对接研究中产生的顶部撞击复合物,使用Schrodinger Desmond MD模拟包对游离蛋白和所有三个顶部撞击复合物进行了100 ns的模拟。自由HTN RdRp的MD模拟显示,初始均方根偏差为3 ?,在30 ns模拟时间内增加到5.4 ?。然后下降到4.0 ?,超过这个数值,它再次上升,从50到100秒,它在5.4 ?左右。因此,在50 - 100 ns的一半模拟时间内,它处于平衡状态,波动较小(图3)。均方根波动表明,HTN RdRp内切酶的初始20个氨基酸和残基数在80 - 90之间波动最大,而其余氨基酸在HTN RdRp内切酶中是相当刚性的(补充图3)。
图3
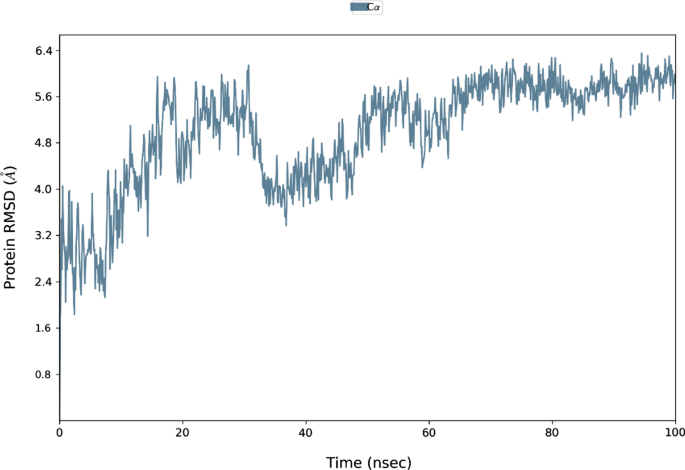
HTN RdRp内切酶在100 ns模拟时间内的C-α均方根偏差
正如之前在分子对接研究中观察到的那样,顶部确定的命中与HTN病毒内切酶结构域的Mn2+离子和其他活性位点残基有效相互作用,在MD模拟相互作用研究中也是如此,这些配体在100 ns MD模拟时间内与靶蛋白活性位点有效相互作用。这些相互作用沿着所有模拟复合物的模拟轨迹显著发生。在100 ns模拟时间内,由于与配体的络合作用,蛋白质与配体络合的RMSD表现出不同的行为(图4a)。在30、40和80 ns模拟时间窗内,蛋白质的RMSD从最初的2.5 ?持续波动,然后上升。在45秒左右,它甚至跃升到7.5 ?,但在模拟时间结束时,蛋白质的RMSD为5.5 ?。在图4a中,栗色代表了在蛋白质结合袋内的ingavirin配体的RMSD。可以观察到,在模拟时间内,由于配体与蛋白质之间的各种接触的建立和破坏,配体的RMSD也会出现波动。配体的平均RMSD在4-5 ?范围内(图4a)。配体复合物中蛋白质C-α氨基酸的RMSF波动与游离蛋白有很大不同(见图3)。在与ingavirin复合物中,HTN RdRp的前20个氨基酸的RMSF波动高达9 ?,而80-90氨基酸区域的RMSF波动较小。这些残基的这种低运动是与英格韦林接触的各种残基的间接影响(图4a右图)。
图4
a左图显示蛋白质的RMSD(蓝色),而栗色是配体的RMSD,配体是HTN RdRp内切酶复合物的一部分。右图显示了与配体复合物的蛋白质氨基酸残基的RMSF。b在MD模拟过程中Ingavirin与htnrdrp核酸内切酶结构域的二维接触图。c相互作用分数图或由活性位点的相互作用氨基酸产生的相互作用类型。d HTN病毒核酸内切酶结构域活性位点的相互作用氨基酸沿100 ns模拟轨迹与ingavirin相互作用
通过对ingavirin与HTN RdRp复合物的MD模拟可以观察到(图4b), ingavirin通过离子相互作用与HTN内切酶活性位点的锰离子有效相互作用,并与配合这些离子的其他活性位点残基相互作用。Ingavirin还通过这些Mn2+离子与活性位点的Asp40、Glu110、Asp97和Glu54氨基酸相互作用。
此外,含有氮原子的ingavirin杂环也与HTN病毒核酸内切酶结构域的Asp37形成氢键。此外,在100ns MD模拟过程中,这些涉及Mn2+离子的离子相互作用出现在60%以上的可能性。从MD模拟结果可以推断,似乎英格韦林羰基和羟基是与HTN病毒核酸内切酶结构域相互作用所必需的。在整个模拟过程中,ingavirin + HTN核酸内切酶结构域接触的相互作用比例如图4c所示,HTN病毒核酸内切酶结构域活性位点之间的离子相互作用是主要的相互作用类型。Asp40, Glu54, Asp97和Glu110可以看到它们与活性位点的锰离子进行了显著的离子相互作用。跨水分子桥的接触尤其值得注意,例如与Asp40、Glu54、Asp97和Glu110的水辅助离子接触。在图4d中,沿100 ns模拟轨迹可以看到HTN病毒内切酶结构域相互作用氨基酸的强度,这表明在MD模拟过程中,ingavirin与靶蛋白稳定结合。补充图4显示了在100 ns模拟过程中,蛋白与ingavirin复合物的各种表面积参数、旋转半径和RMSD的变化。
通过Schr?dinger Desmond MD模拟软件模拟Peramivir和htnrdrp核酸内切酶结构域复合物100 ns。图5a(左面板)显示了RdRp内切酶的RMSD(蓝色)。可以观察到,在10 ns的模拟时间内,蛋白质的RMSD从1.8上升到5.6 ?,然后保持不变,直到40 ns有较小的波动。蛋白质的RMSD在40 ns时从4 ?上升到70 ns,然后在4.8 ?的范围内波动,直到模拟时间结束。蛋白质-配体复合物中存在的配体的RMSD(栗色)的初始值为1.6 ?,略有上升,到40 ns的平均值为2.4 ?,但从40 ns突然上升到4 ?,平均到60 ns。从70到100 ns模拟时间窗,观察到进一步的上升,平均值为13-14 ?。较高的RMSD表明蛋白质与配体(peramivir)之间的接触较少。图5a右侧为HTN RdRp内切酶与peramivir复合物中氨基酸残基c - α的均方根波动。初始5-20个氨基酸的RMSF最大值为4.8 ?。而氨基酸80-90的蛋白质区域在模拟时间内表现出3.2 ?的均方根波动,而蛋白质中其余的氨基酸在模拟时间内是相当刚性的。
图5
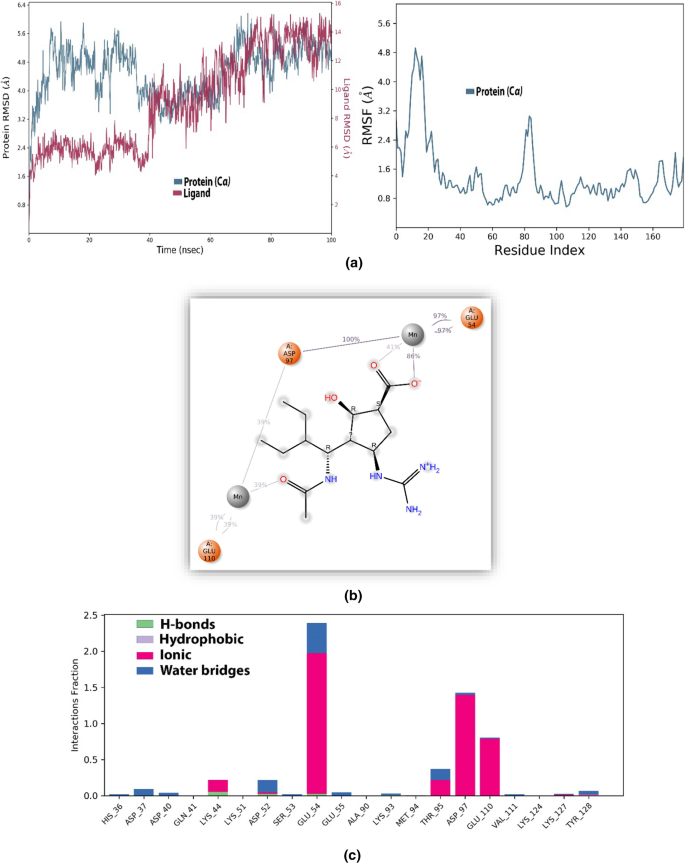
a左图显示蛋白质的RMSD(蓝色),而栗色是配体的RMSD,它是HTN RdRp内切酶复合物的一部分。右图显示了与配体复合物的蛋白质氨基酸残基的RMSF。b在MD模拟过程中,帕拉米韦与htnrdrp核酸内切酶结构域的二维接触图。c HTN RdRp与Peramivir相互作用氨基酸的相互作用分数图或相互作用类型
在peramivir和HTN RdRp复合物MD模拟评估中,peramivir通过离子相互作用有效地与HTN内切酶活性位点的锰离子相互作用,以及与配合这些离子的其他活性位点残基相互作用,如图5b所示。活性位点的两个锰离子都与帕拉米韦配体有很强的相互作用。该配体的羧基部分主要与靶蛋白的一个Mn+2离子相互作用。该羧基的羟基和羰基分别与Mn++离子接触,羟基-OH与Mn+2离子接触的时间超过100 ns模拟时间的80%。相反,羧基的羰基与活性位点的Mn+2离子相互作用的时间超过了模拟时间的40%。该羧基还通过Mn+2离子与活性位点的Glu54和Asp97氨基酸相互作用(图5b)。帕拉米韦的乙酰胺基羰基氧原子与活性位点的第二锰离子相互作用。这个乙酰胺的羰基氧原子也与HTN RdRp核酸内切酶结构域活性位点的Glu110和Asp110残基相互作用。
该配合物在100 ns模拟过程中的相互作用分数图如图5c所示,配体peramivir与HTN RdRp核酸内切酶结构域的锰离子和其他活性位点残基存在显著且持续的相互作用。我们的配体peramivir与HTN RdRp核酸内切酶结构域的Glu54、Thr95、Asp97和Glu110发生了多种类型的相互作用,主要是离子相互作用。像跨水分子桥的接触,如帕拉米韦水辅助离子接触到HTN RdRp核酸内切酶结构域的Asp40、Glu54、Asp97、Thr95和Glu110活性位点残基,是特别有意义的相互作用。在100 ns模拟轨迹中,帕拉米韦与活性位点的相互作用是显著的相互作用,在MD模拟过程中配体与靶蛋白接触,表明帕拉米韦与靶蛋白的结合是稳定的。补充图5显示了在100 ns模拟过程中,蛋白质与peramivir复合物的各种表面积参数、旋转半径和RMSD的变化。
我们研究的第三个确定的先导化合物的MD模拟是2 ' -脱氧-2 ' -氟胞苷。虽然该化合物在分子对接研究中与靶蛋白表现出明显的相互作用,但是,当模拟该配体-蛋白复合物100 ns时,我们注意到该配体在模拟中结合不是很稳定,在MD模拟研究中与靶蛋白相互作用的次数很少。虽然在本计算研究中,我们单独考虑HTN RdRp内切酶,并与小分子抑制剂进行对接和模拟。然而,在生理条件下,不同的核衣壳、核蛋白和RNA分子与汉滩病毒的RdRp持续接触[43]。这些蛋白主要保护和稳定聚合酶,对酶发挥变构作用,并通过为RdRp提供不同的反应分子来增强催化活性[44]。此外,在病毒RNA活性的合成过程中,这种多任务酶处于持续的动态状态,其催化位点对模仿产生RNA分子的核苷酸分子的药物抑制剂是暂时可用的。
在这项研究中鉴定的化合物先前已被证明对多种病毒具有广谱抗病毒活性。例如,本研究发现一种先导化合物ingavirin对人偏肺病毒(hMPV)具有抑制活性。hMPV是一种呼吸道病毒,可引起上呼吸道和下呼吸道感染。研究表明,在50 ~ 500 μg/mL浓度下,ingavirin可有效阻断该病毒的复制[45]。对另一种呼吸道病原体副流感病毒(PI)的研究表明,英格韦林能有效抑制该病毒,保护支气管上皮,并通过降低PI病毒引起的细胞病变作用发挥其抑制活性[46]。同样,对同样是一种致病性呼吸道病毒的人腺病毒(5型)的研究表明,英格韦林也可以通过在感染过程中干扰该病毒的正常形态发生来抑制该病毒[47],与这些病毒一起,英格韦林也是一种抗多种流感病毒(IAV)变体的首选药物,并且是一种被批准用于IAV引起感染的药物[34,48]。研究人员还提出了可能对SARS-CoV-2具有抗病毒活性的Ingavirin及其衍生物[49,50]。
2 ' -脱氧-2 ' -氟胞苷是分子对接研究中发现的另一种铅,也有报道称对克里米亚-刚果出血热病毒(CCHFV)有活性,CCHFV也引起人类出血热,研究表明,2 ' -脱氧-2 ' -氟胞苷比利巴韦林更有效地抑制该病毒的活性,利巴韦林也是一种有效的广谱抗病毒药物[51]。据报道,该化合物的几种衍生物也通过靶向其RdRp酶对HCV具有活性[52]。此外,peramivir也是抗甲型和乙型流感病毒(IAV)的首选药物,文献报道其对几种IAV毒株(H1N1, H5N1)具有广谱活性[53,54]。
计算机辅助药物设计和发现(CADD)技术,如我们在本研究中使用的技术,之前已经成功地识别了针对各种病毒性疾病中几种靶蛋白的有效抑制化合物[31,55,56,57]。因此,考虑到过去的文献研究显示这些顶级化合物对多种人类致病病毒具有充分的抗病毒活性,从本研究中可以推断,进一步的体外和体内细胞实验研究这些化合物对HTN病毒的作用有助于确定这些化合物对HTN病毒引起的肾综合征出血热(HFRS)和汉坦病毒心肺综合征(HCPS)的效力。
ccDownload: /内容/ pdf / 10.1007 / s11030 - 022 - 10567 - 6. - pdf
点击分享到









